吉林國際注冊eCTD
電子遞交的合規性與FENG險管理歐盟要求申請人確保電子資料與紙質版本完全一致,若未在規定時間提交紙質文件可能導致注冊終止。驗證過程中,“錯誤”級別問題(如文件命名不規范、XML邏輯錯誤)必須修正,而“警告”和“提示信息”則建議優化以提升審評體驗。EDQM和EMA均提供驗證工具,申請人需在遞交前完成內部預驗證。G方費用結構與支付流程歐盟eCTD遞交費用因審評程序類型而異:集中程序費用較高,涵蓋科學評估和合規審查成本;G家程序費用由各成員國自行設定。CEP申請需向EDQM支付評審費,具體金額根據原料YAO類型和變更復雜度分級。繳費需通過G方指定渠道完成,并附上付款憑證作為模塊1的組成部分。多語言支持與翻譯要求盡管歐盟允許使用英語提交,但部分成員國要求模塊一的行政文件翻譯為本地語言。臨床試驗數據庫(如SDTM和ADaM)需以英語呈現,同時提供雙語標簽以支持多國審閱。Z業翻譯服務在確保技術術語準確性方面至關重要,尤其針對復雜YAO學和非臨床數據。瑞士IND注冊申報相關技術支持。吉林國際注冊eCTD
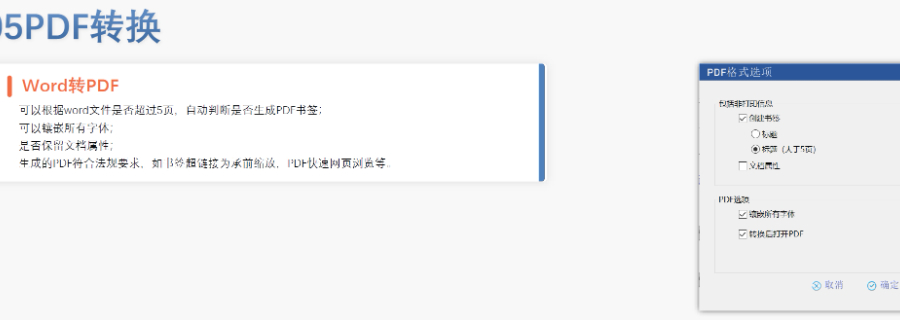
仿制yao作為提高yao物可及性與可負擔性的一類yao物,2012年以前,注冊審評是不收取任何費用的,但當時仿制yao申請積壓嚴重,從申報到獲批需要3~5年的時間。美國國會于2012年頒布了仿制yao使用者費用修正案(GenericDrugUserFeeAmendments,GDUFA),該法律要求制yao行業支付一定的用戶費用,以補充仿制yao申請的審評以及現場檢查的費用,減少仿制藥申請積壓,縮短審評時間,增加基于風險的現場檢查等,其目的是加快公眾獲得安全you效的仿制yao,并降低行業成本。GDUFA必須每五年重授權一次,于2017年更(GDUFAII),于2022年更(GDUFAIII);目前收費種類分為以下四種:ANDA審評費、DMF審評費,在審評時一次性繳納;項目費(Programfee)、設施費(Facilityfee),是上市后每年繳納一次。 甘肅國內注冊eCTD中NDA注冊申報相關技術支持。
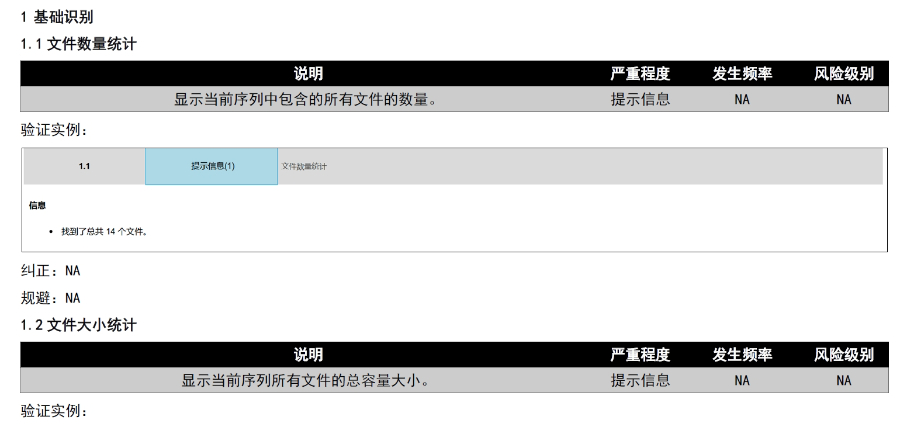
美國電子提交通道ESG(ElectronicSubmissionsGateway)是美國食品yao品監督管理局(FDA)建立的電子化監管信息提交系統,旨在為制yao、生wu制品、醫療器械等行業提供安全、gao效的電子申報服務。自2006年啟用以來,ESG已成為FDA接收電子監管材料的入口,每日處理上千份提交文件,涵蓋上市前審批、上市后監管、臨床試驗數據、不良反應報告等多種類型。該系統通過數字證shu加密和公鑰基礎設施(PKI)技術,確保文件傳輸的真實性、完整性和不可否認性,符合FDA對電子提交的嚴格合規要求。在技術層面,ESG具備強大的文件處理能力。2018年系統升級后,取消了單個文件8GB的限制,可支持高達35GB的大型文件提交,進一步滿足復雜申報需求。此外,文件格式需遵循eCTD(電子通用技術文檔)規范,包括模塊化結構、PDF標準化和XML元數據整合,以確保全球監管機構兼容性。2025年3月28日起,FDA將啟用新一代平臺ESGNextGen,逐步替代現有系統,過渡期需關注兼容性和穩定性問題。
eCTD的實施為監管機構和企業帶來了多重機遇。電子化申報資料能夠極大地加速審評效率,減少人為判斷錯誤和數據混淆的情況,從而提高審評的準確性和速度。同時,eCTD帶來的數據標準化機遇使得全球監管機構的資料內容和電子格式得以統一,有助于在不同監管機構之間進行數據傳輸和共享。這對于提升全球監管效率和行業研發效率具有重要意義。此外,eCTD的實施還促進了全球合作,構建了全球監管的底層大數據基礎。對于企業而言,eCTD提供了一個規范化的研發活動模板,有助于降低與監管機構溝通的成本,提高申報效率。特別是對于國內的醫技術企業而言,eCTD的實施更是具有重要意義,有助于這些企業更好地走向全球市場。然而,中小企業在享受這些機遇的同時,也面臨著技術和成本壓力。eCTD的實施需要專門的團隊進行系統維護和開發,這對于中小企業來說是一筆不小的開支。同時,數據安全問題也是企業關注的焦點。此次CDE擴大eCTD實施范圍對行業而言是一個積極的風向標。短期內,企業面臨的挑戰包括適應更高要求的技術規范并提高文件質量、和eCTD出版系統的磨合以及進行eCTD知識的跨職能培訓等。中ANDA注冊申報相關技術支持。
歐洲YAO品管理局:集中審評程序由歐洲YAO品管理局(EuropeanMedicinesAgency,EMA)負責協調。人用YAO品委YUAN會:人用YAO品委YUAN會(CommitteeforMedicinalProductsforHumanUse,CHMP)負責提供科學意見。歐盟委員會:CHMP的意見隨后被提交給歐盟委YUAN會(EuropeanCommission,EC),由歐盟委會做出是否授權的終決定。這個決定在整個歐盟都是具有法律約束力的。審批過程:申請人向EMA提交申請,包括eCTD(電子通用技術文檔)格式的YAO品注冊文檔。EMA的CHMP分配一個科學評估團隊(Rapporteur和Co-Rapporteur),負責初步評估。CHMP基于評估團隊的報告提供科學意見。歐盟委會根據CHMP的意見做出終決定,批準或拒絕YAO品上市。授權范圍如果YAO品獲得批準,將獲得在整個歐盟、冰島、列支敦士登和挪威YOU效的上市許可(CentralMarketingAuthorisation,CMA)。 歐盟eCTD注冊外包相關技術支持。高新區化學藥品eCTD性價比
eCTD注冊咨詢相關技術支持。吉林國際注冊eCTD
eCTD的法規框架與技術規范:歐盟eCTD的fa規層級包括指南(Guidelines)、指令(Directive)和fa規(Regulation)。其中,fa規(如CTR)具有直接fa律效力,而指南(如ICH eCTD規范)則為技術操作提供參考。eCTD的結構需符合歐盟模塊1規范(DTD 3.0+),包含行政文件(模塊1)、質量數據(模塊3)及臨床研究報告(模塊5)等內容,并通過XML文件實現數據互聯。例如,CEP(歐洲yao典適用性證書)的eCTD申報需單獨構建信封(Envelope)和模塊1,并指定標識符(UUID)以確保技術合規性。吉林國際注冊eCTD
- 楊浦區eCTD品牌 2025-12-20
- 河北仿制藥eCTD 2025-12-20
- 合肥NDAeCTD品牌 2025-12-20
- 吉林國際注冊eCTD 2025-12-20
- 無錫賦悅科技eCTD找哪家 2025-12-20
- 楊浦區國際注冊eCTD醫療科技 2025-12-20
- 徐匯區中國eCTD報價 2025-12-20
- 蘇州新藥eCTD 2025-12-20
- 北京加拿大eCTD 2025-12-20
- 靜安區CDE eCTD品牌 2025-12-20
- 安徽網站搭建解決方案 2025-12-22
- 瑯琊區開放生態集成型網站搭建響應 2025-12-22
- 天津貨代軟件排行榜 2025-12-22
- 特色數字孿生與CIM平臺商家 2025-12-22
- 貴州Agilia再轉印打印機推薦 2025-12-22
- 汕頭手機批發訂貨小程序 2025-12-22
- AI**智能撰寫批量定稿 2025-12-22
- 北京購買實驗室純水機 2025-12-22
- 崇明區附近短視頻運營 2025-12-22
- 鶴壁網絡營銷哪家正規 2025-12-22