-
 山西Vero宿主細胞殘留DNA檢測方案
山西Vero宿主細胞殘留DNA檢測方案湖州申科生物的宿主細胞殘留 DNA 檢測技術服務,涵蓋樣品適用性驗證服務:針對具體樣品開展適用性驗證,搭配試劑盒的全面性能驗證報告,包含精密度(重復性、中間精密度)與樣品回收率(準確性)驗證,確保檢測結果準確可信;提供定制化 HCD 分析方法全面性能驗證服...
2025-12-16 -
 江西CHO宿主細胞殘留DNA檢測常見問題
江西CHO宿主細胞殘留DNA檢測常見問題湖州申科生物的產品研發與制造過程遵循 ISO13485 質量管理體系要求開展,并參照 CDE《生物制品質量控制分析方法驗證技術審評一般原則》、《中國藥典》9101 分析方法驗證指導原則及 ICH Q2 (R2)《分析方法驗證指導原則》等法規文件,對 SHE...
2025-12-16 -
 北京疫苗產品用宿主細胞蛋白(HCP)殘留檢測橋接驗證
北京疫苗產品用宿主細胞蛋白(HCP)殘留檢測橋接驗證湖州申科運用免疫磁珠分離技術(IMBS),搭配 2D 電泳或 LC-MS 技術評估抗體覆蓋率。IMBS 的主要流程涵蓋:多克隆抗體與磁珠的偶聯反應、磁珠未結合位點的封閉處理、HCP 樣本與結合抗體的磁珠共同孵育反應。該過程中,HCP 抗體會結合可識別的 H...
2025-12-16 -
 浙江非動物源內毒素檢測動態顯色法鱟試劑
浙江非動物源內毒素檢測動態顯色法鱟試劑低內毒素回收(LER)的主要形成機制之一是螯合劑與非離子表面活性劑的協同作用,直接影響內毒素檢測結果。第一步,樣品中的螯合劑(如檸檬酸鹽)會去除二價陽離子(Mg2?、Ca2?),削弱 LPS 聚集體的鹽橋結構,降低其剛性;第二步,表面活性劑(如吐溫 20)...
2025-12-15 -
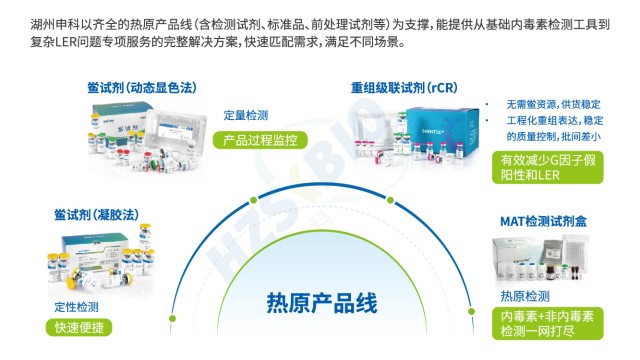 江蘇抗體藥物內毒素檢測技術服務
江蘇抗體藥物內毒素檢測技術服務重組級聯試劑(rCR)通過完整模擬天然鱟試劑的酶促級聯反應路徑,實現高效且特異的內毒素檢測。其反應機制為:內毒素首先活化重組 C 因子,活化的 C 因子進一步活化重組 B 因子,隨后活化重組凝固酶原轉化為凝固酶,再催化顯色底物產生黃色信號(405nm 波長...
2025-12-15 -
江蘇疫苗內毒素檢測重組級聯試劑(rCR)
在進行內毒素檢測時,干擾試驗又叫增強或抑制試驗,主要目的是確證檢測內毒素的方法是否受樣品干擾。在建立細菌內毒素檢查方法中,驗證試驗前,要去除樣品可能含有的內毒素,以確保建立方法的準確可靠。藥典規定:①當進行新藥的內毒素檢查試驗前,或無內毒素檢查項品種建立內...
2025-12-15 -
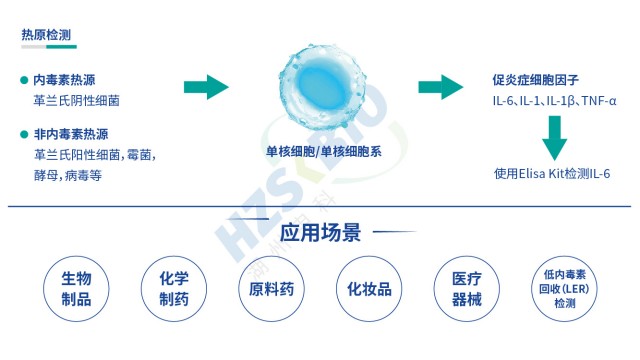 高效熱原檢測法規要求
高效熱原檢測法規要求PyroSHENTEK?熱原檢測試劑盒突破傳統檢測方法局限,不僅能檢測革蘭氏陰性菌來源的內毒素,還可準確識別革蘭氏陽性菌(脂磷壁酸 LTA)、病毒、真菌等產生的非內毒素熱原(NEP),契合熱原檢測 “全風險覆蓋” 的法規需求。其定量限低至 0.025EU/...
2025-12-15 -
 北京內毒素檢測抗干擾方案
北京內毒素檢測抗干擾方案內毒素檢測法規體系正逐步向非動物源試劑傾斜,為重組試劑的應用鋪平道路。美國藥典(USP)已將重組 C 因子(rFC)和重組級聯試劑(rCR)正式收錄,于2025 年 5 月納入 USP-NF,明確要求用戶驗證重組試劑對特定產品的適用性。歐洲藥典(EP)通則...
2025-12-15 -
重慶生物制品宿主細胞殘留DNA檢測常見問題
SHENTEK? CHO 殘留 DNA 檢測試劑盒,可對各類生物制品及藥品的中間品、半成品與成品中殘留的 CHO 宿主細胞 DNA 進行定量檢測。該試劑盒基于熒光探針原理實現對樣品中 CHO 殘留 DNA 的定量,不僅檢測高效快速、專一性突出、性能穩定可靠...
2025-12-12 -
 江蘇內毒素檢測LER現象
江蘇內毒素檢測LER現象內毒素檢測常與熱原檢測混淆,二者既有關聯又有區別:熱原是指所有能引起發熱的物質(包括內毒素、病毒、真菌等),通過傳統家兔熱原試驗檢測;內毒素是熱原的主要成分(占 90% 以上),檢測更具特異性。目前,家兔熱原試驗因操作復雜、動物成本高,已逐漸被單核細胞活化...
2025-12-12 -
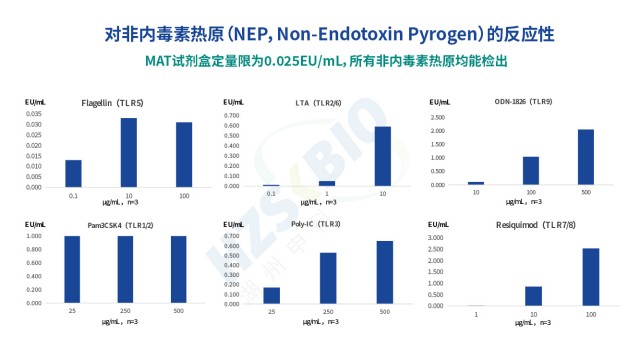 山東疫苗熱原檢測
山東疫苗熱原檢測熱原檢測MAT法的法規地位持續提升,已成為多國藥典認可的熱原檢測替代方法。歐洲藥典(EP)是推動 MAT 應用的關鍵力量:通則 2.6.30 明確 MAT 可替代家兔熱原試驗(RPT),且能同時檢測內毒素與非內毒素熱原;2024 年歐洲藥典委員會批準刪除所...
2025-12-12 -
 上海內毒素檢測技術服務
上海內毒素檢測技術服務β- 葡聚糖是鱟試劑(LAL)檢測內毒素的常見干擾物,可活化 LAL 中的 G 因子通路,導致假陽性結果。干擾多見于含植物源原料的樣品(如中藥注射劑)、生物發酵產物或環境真菌污染的樣品。消除方法包括:使用特異性 LAL 試劑(如添加葡聚糖抑制劑的 LAL)...
2025-12-12 -
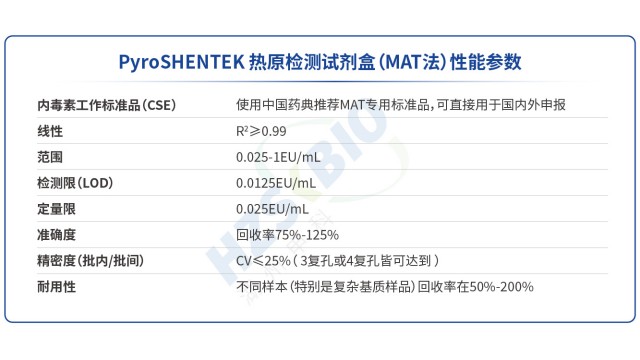 北京合規性熱原檢測MAT試劑盒
北京合規性熱原檢測MAT試劑盒MAT法熱原檢測中,非內毒素熱原(NEP)對照品設為 “選做”,且試劑盒不默認配備,需結合檢測需求靈活使用,背后有明確的設置邏輯。首先,試劑盒開發階段已通過驗證(用多種 NEP 配體刺激細胞),證明其可檢出 NEP,后期實驗是否加入 NEP 對照,只需根據...
2025-12-12 -
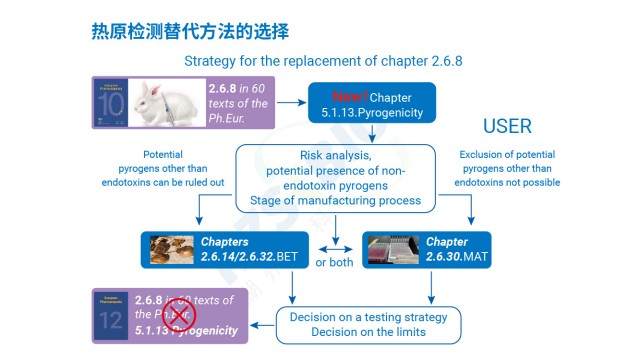 安徽熱原檢測方法驗證
安徽熱原檢測方法驗證歐盟在熱原檢測方法選擇上,以動物保護和檢測準確性為導向,形成明確的法規傾向。首先,歐盟禁止家兔法(PRT 法)這類動物實驗,要求采用替代方法,單核細胞活化試驗(MAT 法)因符合 3R 原則(替代、減少、優化),被納入歐洲藥典(EP2.6.30),成為熱原...
2025-12-12 -
重組蛋白內毒素檢測操作步驟
在為新物料或新產品、中間產品建立細菌內毒素檢測方法時,常會遇到各種困難,尤其是尚處于新藥研發早期階段的藥物。此時,由于藥物制劑、緩沖系統等還不穩定,經常會發生變化,這樣就給方法的建立帶來了不同程度的影響。在建立細菌內毒素檢查法之前,須盡可能多地了解有關該藥...
2025-12-12 -
 安徽E.coli宿主細胞殘留DNA檢測常見問題
安徽E.coli宿主細胞殘留DNA檢測常見問題SHENTEK?E1B殘留DNA檢測試劑盒,可對生物制品中宿主細胞(HEK293及其衍生細胞系,如293T、293F等)的E1B殘留DNA進行定量檢測。該試劑盒基于熒光探針原理,通過qPCR方法實現對樣品中E1B殘留DNA的定量分析,具備檢測快速、專一性強...
2025-12-12 -
 江蘇合規性內毒素檢測
江蘇合規性內毒素檢測SHENTEK?重組級聯試劑為內毒素檢測高效解決方案。法規層面,符合美國藥典(USP)、歐洲藥典(EP)及日本藥典(JP)要求,滿足國際標準檢測需求。抗干擾性強,與天然鱟具有相同反應機制,檢測結果等效,適配復雜樣本場景。特異性表現突出,不含 G 因子,避免...
2025-12-12 -
 廣東Human宿主細胞殘留DNA檢測常用知識
廣東Human宿主細胞殘留DNA檢測常用知識SHENTEK?Hi5&AcNPV殘留DNA檢測試劑盒(多重PCR-熒光探針法),可對昆蟲細胞(HighFive)桿狀病毒表達系統所生產的基因工程疫苗內,殘留的Hi5細胞DNA與桿狀病毒(AcNPV)DNA開展定量檢測。該試劑盒依托熒光探針原理,結合多重q...
2025-12-11 -
 北京疫苗產品支原體檢測驗證菌株
北京疫苗產品支原體檢測驗證菌株培養基的科學選擇與合規使用是支原體培養法檢測成功的基礎,湖州申科按 USP 標準明確了三類推薦培養基的適用場景。Hayflick Media 用于支原體一般性檢測,Frey Media 專門針對滑液囊支原體檢測,Friis Media 則適用于非禽類支原體...
2025-12-11 -
 成都漢遜酵母宿主細胞蛋白(HCP)殘留檢測
成都漢遜酵母宿主細胞蛋白(HCP)殘留檢測依據美國藥典 1132 章節規定,HCPs 校準品需具備代表性,能夠覆蓋實際產品生產工藝中的 HCPs。結合 HCP 免疫檢測方法的使用目的與預期風險管理需求,為滿足工藝開發、驗證需求,同時應對下游工藝可能出現的異常失效或工藝變更情況,建議選用上游發酵工藝...
2025-12-11 -
 成都Sf9宿主細胞蛋白(HCP)殘留檢測
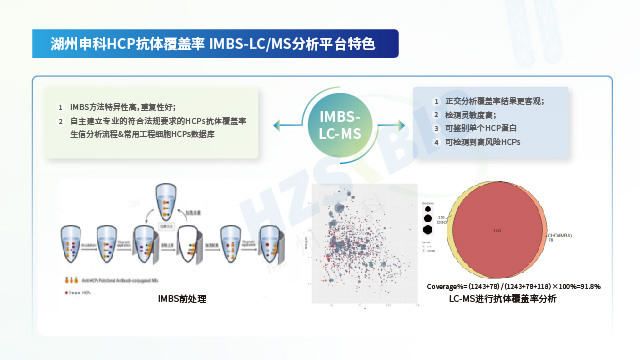
成都Sf9宿主細胞蛋白(HCP)殘留檢測法規推薦的抗體覆蓋率評估方法主要分為兩類:一類是傳統 2D-WB 法,另一類是基于抗體親和的免疫捕獲類方法。傳統 2D-WB 法存在諸多不足,如需對蛋白進行變性處理、樣品需前處理(會破壞蛋白天然表位)、轉膜效率較低、易出現非特異性反應等,因此難以準確反映真...
2025-12-11 -
湖南Vero宿主細胞殘留DNA檢測生產企業
SHENTEK? HEK293 殘留 DNA 檢測試劑盒,可對各類生物制品中間品、半成品及成品中的 HEK293 宿主細胞 DNA 開展定量檢測。該試劑盒依托熒光探針原理實現對樣品中 HEK293 殘留 DNA 的定量,不僅檢測速度快、專一性突出、性能穩定...
2025-12-11 -
浙江醫療器械熱原檢測
中國藥典對 MAT 法熱原檢測要求 4 復孔,未明確 CV 限值,需結合細胞實驗特性合理解讀與操作。藥典不設 CV 限值的主要原因是:MAT 法基于細胞反應,細胞活性易受環境微小變化(如溫度、pH)影響,存在天然不穩定性,過嚴的 CV 要求可能脫離實際;但...
2025-12-11 -
 江蘇合規性內毒素檢測技術升級
江蘇合規性內毒素檢測技術升級當實驗室更換內毒素檢測方法或更換試劑供應商時,需進行方法比對與橋接驗證。比對實驗需選取至少 3批代表性樣品,分別用新舊方法檢測,計算結果相關性(如相關系數 R2≥0.95)和偏差(≤20%)。橋接驗證還需評估新方法的特異性、靈敏度是否與舊方法一致,如確認對...
2025-12-11 -
 江蘇生物制品宿主細胞殘留DNA檢測生產企業
江蘇生物制品宿主細胞殘留DNA檢測生產企業湖州申科生物的產品研發與制造過程遵循 ISO13485 質量管理體系要求開展,并參照 CDE《生物制品質量控制分析方法驗證技術審評一般原則》、《中國藥典》9101 分析方法驗證指導原則及 ICH Q2 (R2)《分析方法驗證指導原則》等法規文件,對 SHE...
2025-12-11 -
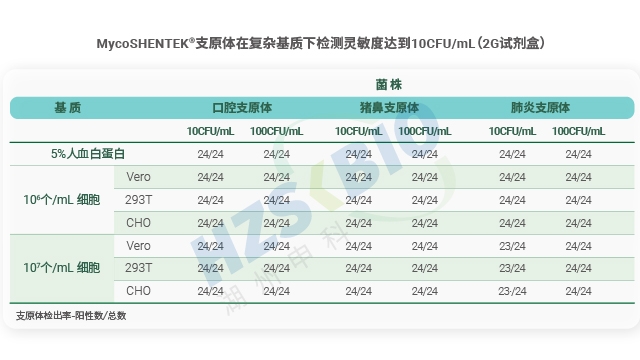
 重慶疫苗產品支原體檢測試劑盒
重慶疫苗產品支原體檢測試劑盒MycoSHENTEK? 支原體 DNA 檢測試劑盒參照 EP 2.6.7 和 JP XVII 支原體檢測相關要求完成全面性能驗證,是經過三方室間驗證、性能完全符合藥典標準的支原體檢測試劑盒。該試劑盒檢測靈敏度高達 10CFU/mL,可同時替代傳統培養法與...
2025-12-11 -
 浙江干細胞產品支原體檢測NAT法
浙江干細胞產品支原體檢測NAT法培養基的科學選擇與合規使用是支原體培養法檢測成功的基礎,湖州申科按 USP 標準明確了三類推薦培養基的適用場景。Hayflick Media 用于支原體一般性檢測,Frey Media 專門針對滑液囊支原體檢測,Friis Media 則適用于非禽類支原體...
2025-12-11 -
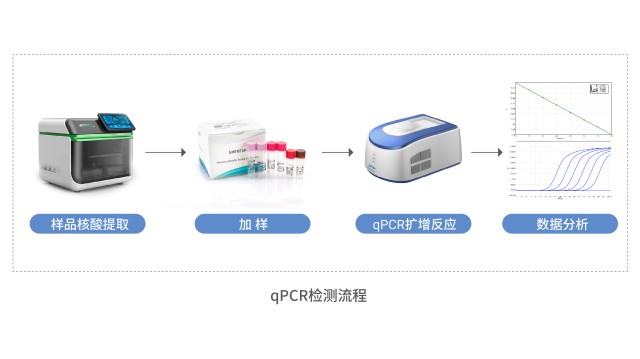
湖南qPCR法宿主細胞殘留DNA檢測方案
生物制品宿主細胞殘留 DNA 檢測是生物制藥領域的關鍵質量控制環節,主要目的是定量監測疫苗、單抗、基因治療產品等生物制品中宿主細胞來源的 DNA 殘留量,以此評估潛在安全風險并確保符合法規要求。其關鍵價值在于:殘留 DNA 可能攜帶致病基因、病毒序列及致瘤...
2025-12-11 -
 湖南細胞療法產品支原體檢測技術服務
湖南細胞療法產品支原體檢測技術服務外源因子全自動核酸檢測分析系統系統在數據追溯與合規管理方面進行了針對性設計,完美適配生物藥行業的嚴格監管要求。系統內置三級權限管理機制,可對操作人員、檢測項目、數據訪問進行準確管控,同時具備完善的日志審計追蹤功能,詳細記錄檢測全流程的關鍵信息,確保數據可追...
2025-12-11 -
重組蛋白熱原檢測合規申報
革蘭氏陽性菌注射劑產品只依賴內毒素檢測存在安全風險,需結合熱原檢測特性制定防控方案。內毒素是革蘭氏陰性菌細胞壁的脂多糖成分,而革蘭氏陽性菌可產生非內毒素熱原(NEPs),如脂磷壁酸,這類物質同樣能引發人體發熱反應,若只檢測內毒素,可能遺漏 NEPs 污染,...
2025-12-11